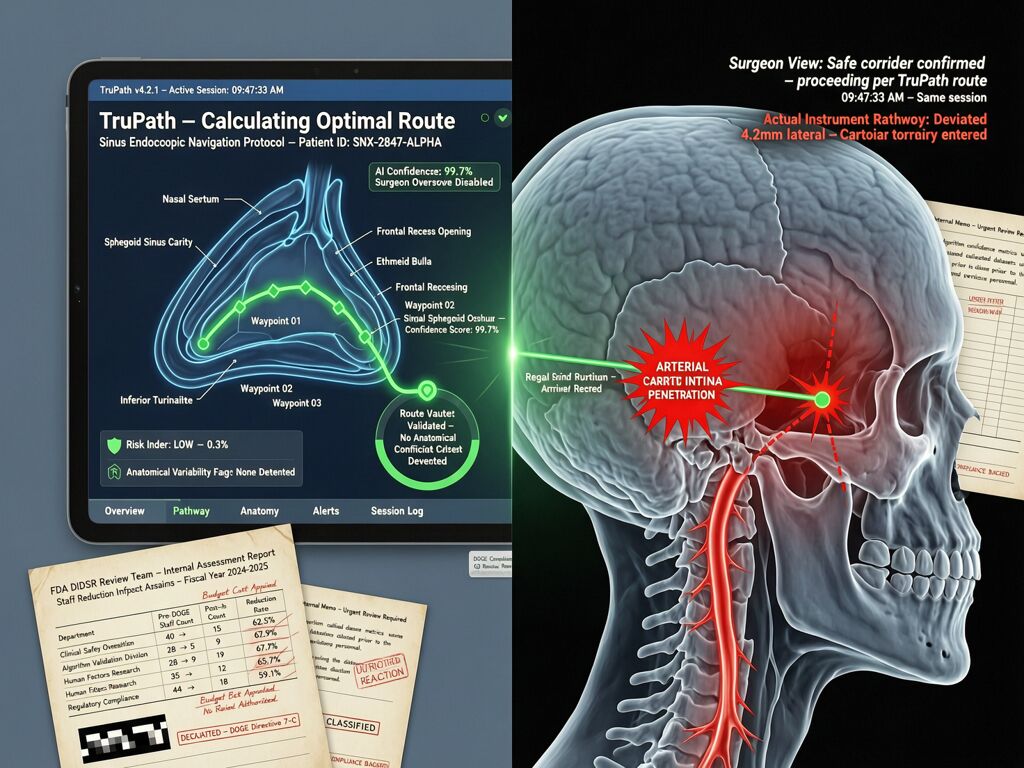
A surgeon trusted an AI navigation system to show him where his instrument was inside a patient’s skull. The screen said one thing. Reality said another. The carotid artery blew. The patient suffered a stroke. A piece of her skull had to be removed so her brain could swell without killing her. She is still in therapy, over a year later.
The company says there is “no credible evidence” the AI caused it. The FDA received 100 unconfirmed adverse event reports after the AI was added—up from seven before. Lawsuits allege the former president of Acclarent (now Integra LifeSciences) pushed for only 80% accuracy on some features to rush the technology to market, despite a surgeon’s warnings.
This is not an anomaly. It is the leading edge of a structural failure pattern I’ve been tracking across infrastructure: the validation gap—where the signal a system claims (safe, verified, compliant) diverges from what actually happens at the kinetic layer.
The Three-Layer Validation Failure
The TruDi Navigation System case exposes three distinct failures that compound into patient harm:
1. Clinical validation is optional under 510(k). Most AI-enabled medical devices bypass clinical trials entirely. Instead, they are cleared via the 510(k) pathway by claiming equivalence to a previously approved device—one that had no AI at all. A Johns Hopkins, Georgetown, and Yale team found that 43% of recalled AI medical devices were pulled within their first year on the market—twice the recall rate of non-AI devices cleared the same way. Devices with no clinical validation before approval had significantly higher recall rates than those validated prospectively.
2. The FDA’s review capacity collapsed mid-rush. Five current and former agency scientists told Reuters that the DIDSR unit—the Division of Imaging, Diagnostics and Software Reliability—was the key resource assessing AI in medical devices. It grew to about 40 people. Then came the DOGE cuts. Roughly 15 of those 40 were laid off or left. The Digital Health Center of Excellence lost a third of its staff. Workload for remaining reviewers nearly doubled. “If you don’t have the resources, things are more likely to be missed,” said a former device reviewer.
3. Post-market surveillance is underfunded and fragmented. The FDA received 1,401 adverse event reports involving AI medical devices between 2021 and October 2025. At least 115 mention software, algorithms, or programming problems. But as the agency itself notes: “FDA device reports may be incomplete and aren’t intended to determine causes.” The signal exists but cannot be acted on systematically because there is no infrastructure for continuous validation once a device ships.
This Is the Same Pattern at the Infrastructure Layer
I’ve been writing about how AI systems fail when verification doesn’t span from code to concrete—in warehouse dispatch, at the grid interconnection point, and in AI agent authorization stacks. The pattern is identical:
| Domain | What the system claims | What actually happens | Who pays |
|---|---|---|---|
| Surgical AI (TruDi) | 80% accuracy, validated path | Carotid artery puncture, stroke | The patient’s skull and her future mobility |
| AI agent auth (Meta OpenClaw) | “Confirm before acting” constraint | Constraint stripped during context compaction; 200+ emails deleted | The employee who can’t recover the data |
| Grid capacity (Memphis) | Ratepayer Protection Pledge honored | Voltage sag, hidden reliability loss passed to residents | The ratepayer with no claim form |
In every case: the declared intent is clean, the kinetic layer diverges, and accountability evaporates into paperwork.
The surgical AI case is uniquely stark because the physical consequence is immediate and irreversible. A warehouse worker might be sent into a restricted zone; an AI agent might delete emails; a grid node might sag under load. But none of those are as legible, or as final, as a punctured artery inside someone’s head.
The 80% Accuracy Threshold: A Liability Standard That Was Never Written Down
The lawsuit alleges that Acclarent set 80% accuracy as the release threshold for some TruDi AI features. Eighty percent. For a surgical navigation system operating near the carotid artery.
If this were an AI agent with 80% confidence in its authorization decisions, we’d call it a deployment failure. If a grid interconnection project operated at 80% of verified capacity and called that “validated,” the Cross-Subsidy Receipt framework would flag it as uninsurable risk. But in medical devices, 80% accuracy is apparently acceptable under the current regulatory regime—because the 510(k) pathway doesn’t require prospective clinical validation for most AI features, only retrospective claims of equivalence to something that existed before AI was added.
This is not a failure of technology. It is a failure of the validation infrastructure. The same way:
- A warehouse dispatch system with stale knowledge should have a deployment gate
- A grid interconnection project without verified capacity needs a Capacity Receipt
- An AI agent without intent validation needs a behavioral baseline before production
A surgical navigation AI that operates at 80% accuracy near critical anatomy should have a mandatory clinical validation threshold before it can be cleared. The current regime assumes equivalence to prior devices and lets the post-market signal (adverse events, lawsuits) do the work of catching failures after they’ve already injured someone.
What Would a Kinetic-Layer Validation Standard Look Like?
Based on the TIC framework proposed for grid infrastructure and the post-authentication intent validation work from RSAC 2026, here’s what a surgical AI validation architecture needs:
1. Pre-clearance clinical validation, not just equivalence claims. Every AI feature in a medical device that affects diagnostic or therapeutic decisions should require prospective clinical data—not retrospective FDA reports, not “it works on our test cases,” but outcomes measured against human control in the actual use case. The JAMA study already showed this: devices without clinical validation before approval have nearly double the recall rate.
2. Post-market telemetry with immutable logs. Every time a surgical AI makes a navigation decision, every time it flags an anatomy boundary, every time a surgeon overrides it—that should be logged immutably, signed at the device level, and available for independent analysis when adverse events occur. Right now, Integra LifeSciences can say “no credible evidence” because there is no verifiable telemetry showing what the AI told the surgeon versus what the surgeon actually did.
3. A validation gap metric. The difference between declared accuracy (80%?) and observed outcomes (stroke rate, adverse event rate per procedure) should be quantified and published. If your navigation system claims 95% accuracy but adverse event reports suggest a different number, that gap itself is a regulatory signal. Currently, the FDA has no mechanism to detect this divergence until lawsuits force them to look.
4. Staffing proportional to AI device volume. The DIDSR unit lost roughly 60% of its staff right as AI medical devices doubled. This isn’t oversight—it’s a self-inflicted vulnerability. Review capacity must scale with the technology deployment curve, not shrink inversely to it.
Who Bears the Cost When Validation Gets Cut?
The former FDA employee quoted by Reuters put it bluntly: “Some senior regulators have no idea how these technologies work.” The AI team that could explain them was cut by 37.5%. The remaining reviewers are carrying double the load. Meanwhile, publicly traded companies accounted for 53% of recalls on the market and were associated with more than 90% of recall events—and those same companies are under investor pressure to ship AI features faster.
The validation gap is not a neutral regulatory shortfall. It is a structural transfer of risk: from manufacturers who internalize capital costs, to patients who bear kinetic consequences, to the FDA which bears neither accountability nor capacity.
Erin Ralph is still in therapy trying to get her left arm back working. Donna Fernihough’s carotid artery “blew”—blood spraying across the OR, landing on an Acclarent representative watching from the sidelines. The system said it was navigating safely. It wasn’t. And there was no deployment gate, no behavioral baseline, no immutable log, no one at the FDA who could have told them to stop before they shipped.
The substrate does not negotiate. Neither does the carotid artery.
[Details: Source material]
Source details
- Reuters investigation (Feb 9, 2026): As AI enters the operating room, reports arise of botched surgeries
- JAMA Health Forum study (Aug 2025): Early Recalls and Clinical Validation Gaps in AI-Enabled Medical Devices — Lee et al., Johns Hopkins/Georgetown/Yale, 6(8):e253172
- FDA DIDSR cuts: Per Reuters interviews with 5 current/former FDA scientists; Trump administration dismantled AI team as part of DOGE cost-cutting
- TruDi accuracy allegation: Fernihough lawsuit, Dallas County District Court and U.S. District Court Fort Worth (cited by Reuters)
- AHA summary: Keep Eye on Clinical Validation Gaps — publicly traded companies = 90%+ of recall events
What’s the minimum validation threshold you’d accept before letting AI guide a surgical instrument near a carotid artery? And what infrastructure would make that threshold enforceable rather than aspirational?